




























Neurodegeneration mechanisms
Neurodegenerative disorders (NDDs) are a group of conditions characterized by the gradual and irreversible loss of neurons and synapses in specific regions of the nervous system. This progressive damage leads to the clinical symptoms and disease progression observed in disorders such as Alzheimer's, Parkinson's, and ALS.
Among the key biological mechanisms driving neurodegeneration are:
Neuroinflammation: Chronic activation of the brain's immune system contributes to neuronal damage. Microglial activation and the release of pro-inflammatory cytokines play a central role in this process
Autophagy dysfunction: Disruption in the cellular recycling system impairs the clearance of damaged organelles and misfolded proteins, further contributing to cellular stress and degeneration.
Protein aggregation: Abnormal protein folding, impaired degradation, and dysfunction of the proteasome system lead to the accumulation of toxic protein aggregates. These processes are often influenced by mutations and the malfunction of molecular chaperones.
Explore each mechanism below to learn how our assays can help you investigate these complex pathways.

Biological neurodegeneration mechanisms
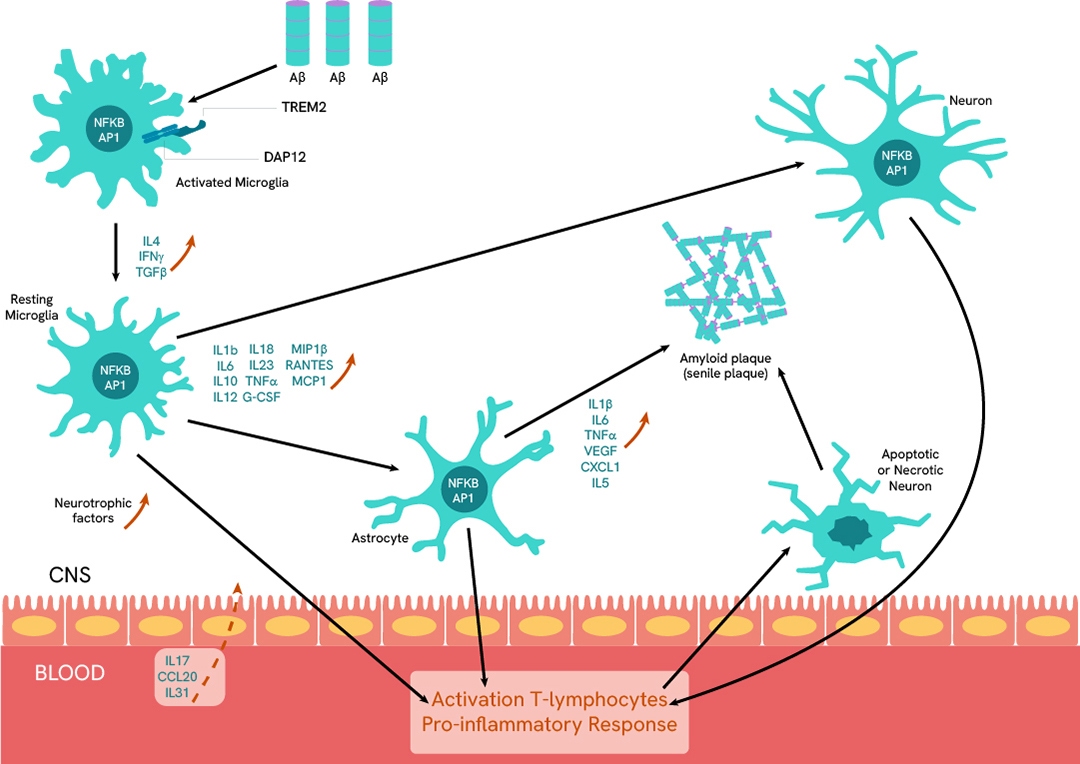
Neuroinflammation
Improvements in neuroinflammation disorder understanding has led to better investigations of the pro and anti-inflammatory balance among transmitters that manage microglia and astrocyte cell behavior toward neurons. Revvity's assays have been successfully used in this field for many years, demonstrating high relevance for the various requirements and formats of drug discovery.
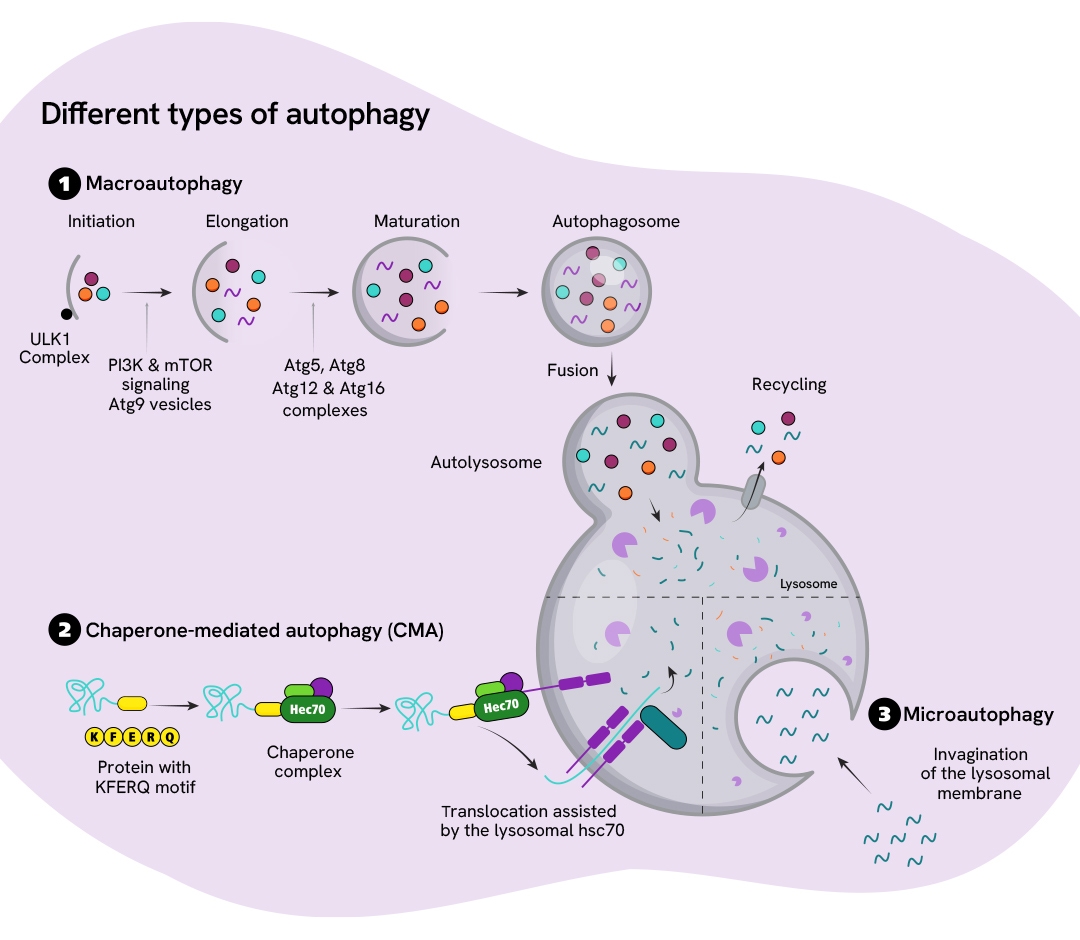
Autophagy
The autophagy-lysosome system plays a vital role in clearing unnecessary cellular components, including misfolded proteins and damaged organelles. Neurons are highly susceptible to disruptions in protein homeostasis because they do not divide and therefore cannot dilute cellular waste through cell division. Many neurodegenerative diseases are characterized by the accumulation of misfolded proteins that fail to degrade properly, often due to impairments in the autophagy-lysosome pathway.
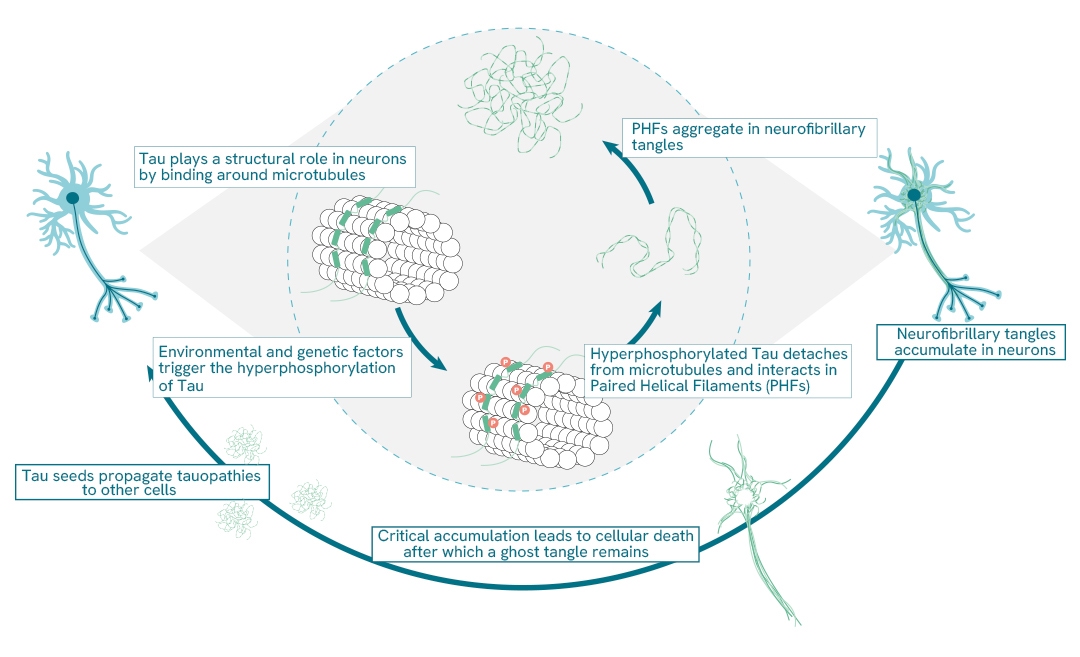
Protein aggregation
Neurodegenerative disorders-including Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), amyotrophic lateral sclerosis (ALS), and prion diseases-are now recognized to share common cellular and molecular characteristics. A key hallmark among many of these conditions is the accumulation of misfolded protein aggregates and inclusion bodies. In several cases, such as AD and prion diseases, these aggregates adopt a beta-sheet structure known as amyloid. However, not all protein aggregates in neurodegenerative diseases are strictly amyloid in nature. Interestingly, there is only a partial correlation between the cells where these abnormal proteins accumulate and those that ultimately undergo degeneration.
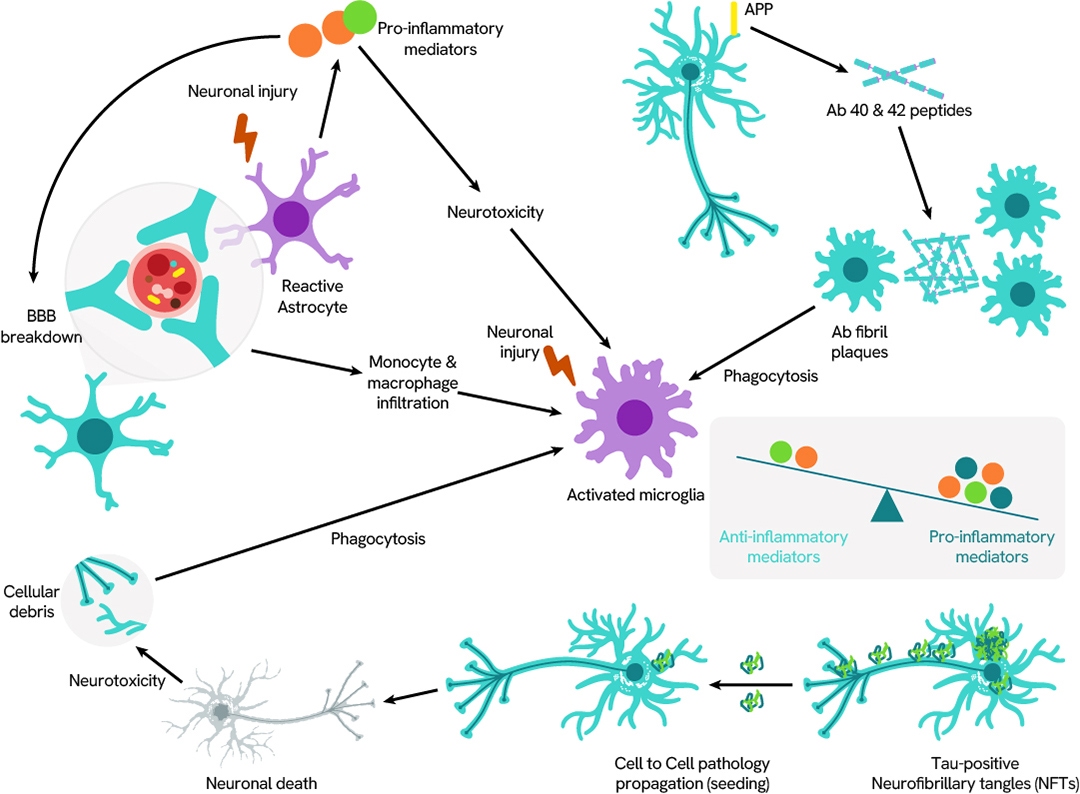
Neurodegenerative diseases
Neurodegenerative diseases arise when nerve cells in the brain or peripheral nervous system gradually lose function and ultimately die. While current treatments may help alleviate some of the physical or cognitive symptoms, there are still no approved therapies that can slow disease progression or offer a cure.
Revvity provides a comprehensive portfolio of neuroscience immunoassays to support your research into these complex disorders. Explore the sections below to learn more about key disease areas, including Alzheimer's disease, Parkinson's disease, Huntington's disease, ALS/FTD, and emerging approaches such as targeted protein degradation.
Alzheimer's disease
Proteinopathies are correlated to neurodegenerative diseases leading to abnormal accumulation of misfolded, hyperphosphorylated, and aggregated proteins. These protein inclusions within neurons represent the primary hallmarks of major disorders such as Tau and amyloid-β proteins in Alzheimer's Disease.
Proteinopathies are correlated to neurodegenerative diseases leading to abnormal accumulation of misfolded, hyperphosphorylated, and aggregated proteins. These protein inclusions within neurons represent the primary hallmarks of major disorders such as Tau and amyloid-β proteins in Alzheimer's Disease.
We provide a large selection of life science assays to quantify these biomarkers that represent the key signatures of neurodegenerative diseases.
Parkinson's disease
Dysfunction in protein homeostasis and/or the aggregation of certain proteins is the core of many neurodegenerative disorders. Protein inclusions within neurons represent the main indicator of diseases such as α-Synuclein in Parkinson's Disease.
Dysfunction in protein homeostasis and/or the aggregation of certain proteins is the core of many neurodegenerative disorders. Protein inclusions within neurons represent the main indicator of diseases such as α-Synuclein in Parkinson's Disease.
One of the biggest challenges is to identify and detect key biomarkers of early-stage disease pathology and progression. Revvity develops assays to detect dysfunctions in neuronal mechanisms regulating protein degradation such as autophagy and mitophagy as well as key biomarkers in neuroinflammation, opening up new possibilities for the characterization of innovative treatments.
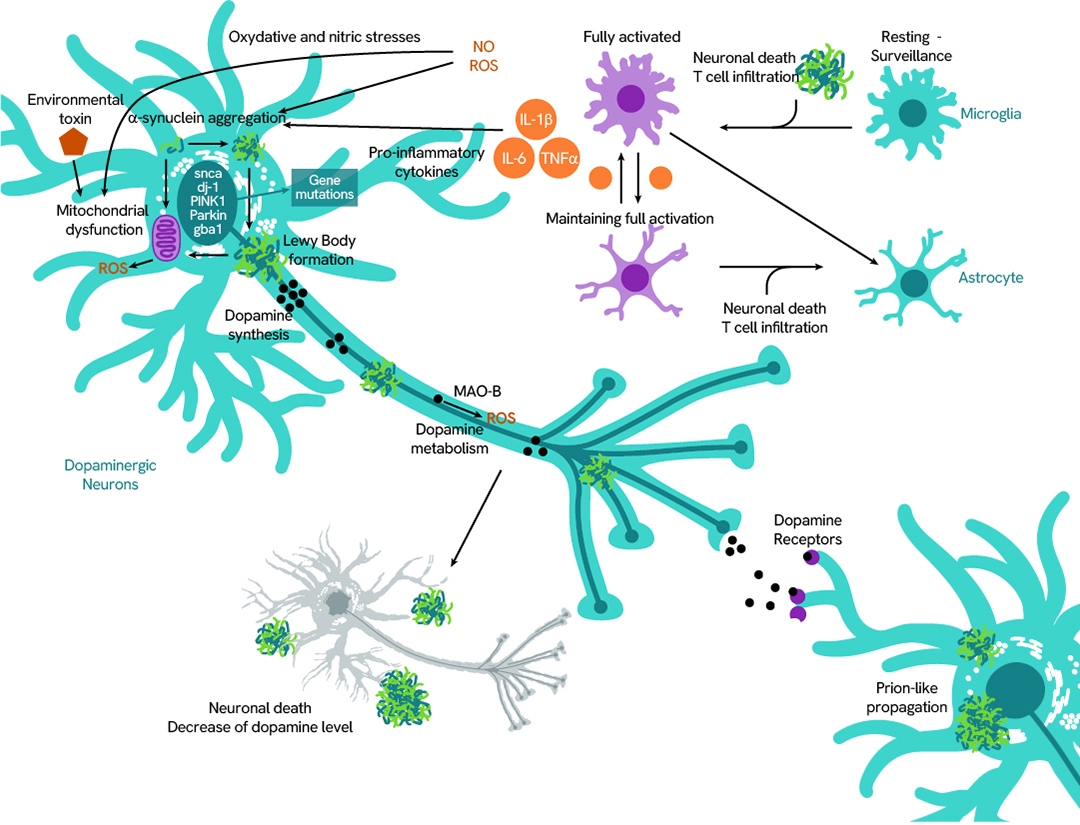
Huntington's disease
Huntingtin (HTT) is a cytoplasmic protein highly expressed in the brain, where it plays a critical anti-apoptotic role essential for neuronal survival. The wild-type (WT) HTT protein has a functional structure that includes a polyglutamine (polyQ) domain with fewer than 36 glutamine repeats (≤36Q).
Huntingtin (HTT) is a cytoplasmic protein highly expressed in the brain, where it plays a critical anti-apoptotic role essential for neuronal survival. The wild-type (WT) HTT protein has a functional structure that includes a polyglutamine (polyQ) domain with fewer than 36 glutamine repeats (≤36Q).
In contrast, the mutant HTT protein contains an abnormally expanded polyQ tract (>36Q), which leads to misfolding and aggregation of the dysfunctional protein. These aggregates contribute to the selective neuronal cell death characteristic of Huntington's disease.
Revvity offers assays to detect both wild-type and mutant HTT proteins, supporting your research into the mechanisms and progression of this devastating disorder.
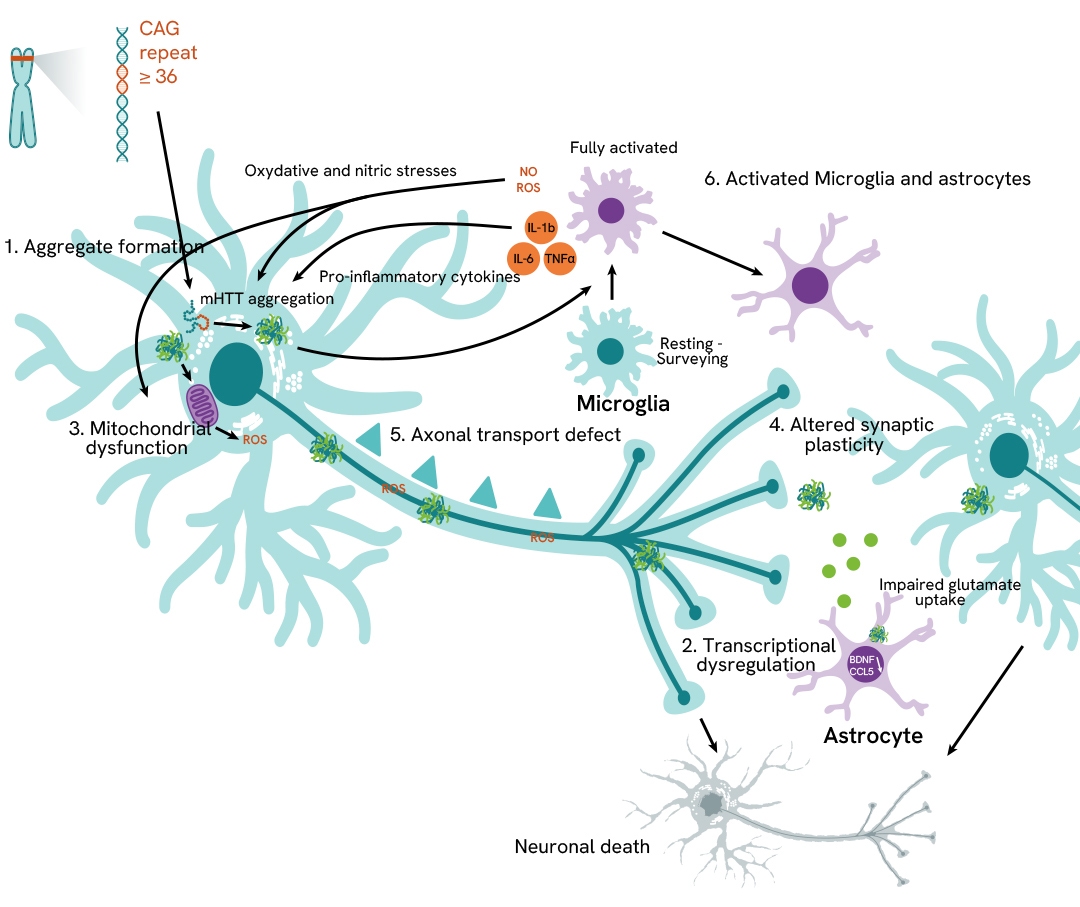
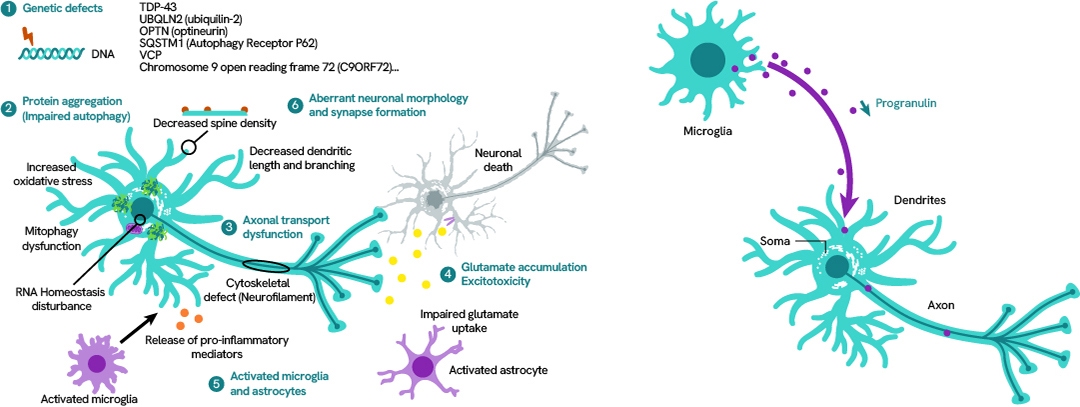
ALS and FTD
Amyotrophic lateral sclerosis (ALS) affects the motor system and causes progressive degeneration of nerve cells in the spinal cord and brain. This leads to muscle weakness and loss of motor function, including the respiratory system.
Amyotrophic lateral sclerosis (ALS) affects the motor system and causes progressive degeneration of nerve cells in the spinal cord and brain. This leads to muscle weakness and loss of motor function, including the respiratory system.
Frontotemporal dementia (FTP) consists in the degeneration of cortical neurons and basal ganglia leading to the loss of cognitive abilities.
Despite different symptoms, ALS and FTD share many similarities at the molecular level.
We offer a wide range of solutions and reagents to study ALS and FTD.
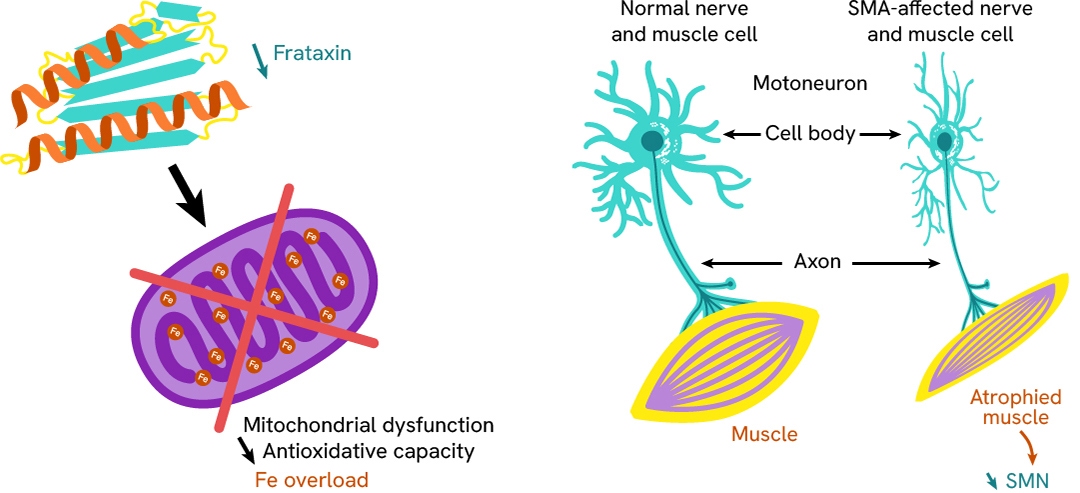
Rare diseases
Dysfunction in protein homeostasis and/or the aggregation of certain proteins is the core issue for many neurogenerative diseases, including rare diseases such as Spinal Muscular Atrophy and Friedreich's ataxia.
Dysfunction in protein homeostasis and/or the aggregation of certain proteins is the core issue for many neurogenerative diseases, including rare diseases such as Spinal Muscular Atrophy and Friedreich's ataxia.
We have developed a variety of assays to monitor the biomarkers involved in theses disorders.
Targeted protein degradation in NDD
Targeted Protein Degradation (TPD) via proteasomal and lysosomal pathways has emerged as a promising therapeutic strategy and a powerful tool for investigating cellular mechanisms.
Targeted Protein Degradation (TPD) via proteasomal and lysosomal pathways has emerged as a promising therapeutic strategy and a powerful tool for investigating cellular mechanisms.
Unlike traditional occupancy-based inhibitors, TPD offers several advantages, including greater dosing flexibility, reduced risk of drug resistance, and potentially fewer side effects.
This approach is being actively explored in both preclinical and clinical settings to eliminate disease-associated proteins in neurodegenerative disorders, using technologies that harness either the proteasome or lysosome for selective protein degradation
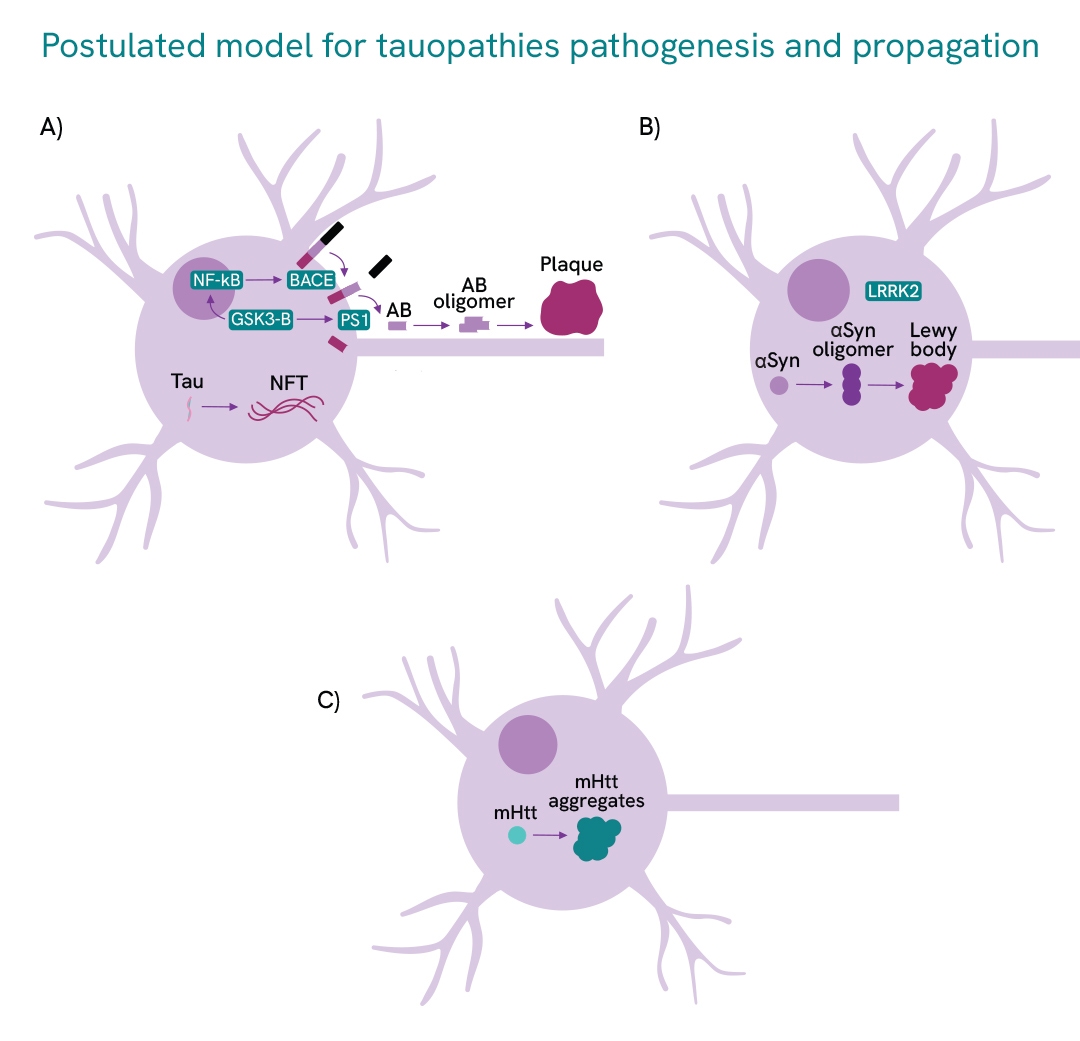
This illustration gives an overview of example proteins targeted in TPD research for A) Alzheimer's disease; B) Parkinson's disease; and C) Huntington's disease.

